
Kevin Karplus
21 February 1995
This paper describes a technique for using models suitable for data compression as a tool for finding interesting sequences in a database. The technique does not require that the structure or consensus sequence for the target sequences be known in advance--only that one or more example sequences be provided. The method can be used for finding repeated elements in a database by providing the entire database as the example sequences.
This paper presents an efficient algorithm for the search, methods for constructing the models automatically, and results of finding clusters of Repetitive Extragenic Palindromic sequences (REPs) in the E. coli genome database, EcoSeq6 [13]. It compares the set of REPs found with previous lists [10,5,14]. The new search techniques do almost as well as the best of previous techniques (self-BLAST), finding about 90% of the known REPs with very strict significance tests (expected number of sequences returned by chance < 0.01) and 92% if the strictness is relaxed.
Although the program works for sequences over any alphabet, only results on DNA sequences are presented here, and some of the techniques (such as complement blurring) are only applicable to nucleic acid sequences.
The approach used here is to define the interesting sequences by using a model m that assigns probabilities to sequences: Pm(s). A sequence is interesting if the probability assigned by the model is significantly higher than the probability of the sequence using a null model. Normally we use the negative log probability of the sequence as a cost measure, since the probabilities become extremely small for longer sequences. Using base-2 logarithms gives us the encoding cost in bits of sequence s using model m:
![]()
Section 2 will describe how the models are constructed, but first let's discuss how they are used for searching efficiently.
We want a search technique whose execution time is linear in d and which returns only disjoint sequences. First, let's assume that the model used to compute the probabilities provides not just an overall cost for a sequence, but assigns a cost to each position of the sequence. Second, let's assume that the cost of any given sequence is the sum of the costs for each of the characters of the sequence. Section 3 discusses the ways in which the models actually used violate these assumptions and why the violations are not serious in this context.
With the above assumptions, we can compute the costs for each character of the database once and save the cumulative costs in an array, called a cost map. From the cost map we can compute the cost for any subsequence of the database by taking the difference between the costs at the end and the beginning of the subsequence.
Hence, given a cost map, finding interesting sequences becomes independent of the model used to create the cost map. This separation of the model from the search technique is one of the main advantages of the techniques described in this paper, allowing a wide variety of model designs. Most other search techniques are firmly tied to a particular model, and often have a zero-order Markov model hard-wired as the null model.
Our problem is to find non-overlapping subsequences of a cost map whose cost is significantly lower than what we would expect from random strings of characters. Furthermore, we would like to do this in time proportional to the number of positions in the cost map.
There are many ways to determine when the cost is significantly lower. The current program uses Milosavljevic's algorithmic significance method [11], which has a simple threshold test to determine if a sequence s is significant:
![]()
![]()
If we want a probability 0.01 or less of getting a sequence
returned due to chance matching, then the threshold is computed as ![]() .In the searching technique described in Section 1.3, one
sequence is checked for each position in the database, so N is the
length of the database d.
For EcoSeq6, with 1,875,932 bases, finding a sequence with
.In the searching technique described in Section 1.3, one
sequence is checked for each position in the database, so N is the
length of the database d.
For EcoSeq6, with 1,875,932 bases, finding a sequence with ![]() requires that its cost under the model be 27.483 bits less than
the null model cost.
requires that its cost under the model be 27.483 bits less than
the null model cost.
This simple threshold technique is easy to implement, and provides a strong test of statistical significance, at least as good as that used for tests of significance in BLAST [1].
This statistical test (like most used in sequence searching) suffers from one major flaw--the database is not really drawn randomly from the distribution implied by the null model. So even if we can refute the null model with high confidence, we have not necessarily found a good match to our model. For example, if a model assigns cost slightly less than 2 bits/base to G and C in most contexts, while the null model assigns 2 bits/base, then almost any sufficiently large GC-rich region would have significant savings. These problems (statistical significance not being equivalent to biological significance and low-entropy regions providing false positives) are well-known, and apply to all search methods [3].
The significance test used here is a very stringent one, so that a short REP cluster in E. coli with only 24 bases would need to be encodable in less than 48-27.479 = 20.521 bits (0.855 bits/base) in order to be found. This stringent condition is difficult to meet with the simple models described in Section 2. Luckily, the regions around the REP clusters are fairly similar, and so the model can usually find a significant, slightly larger region around a REP.
In general, using the strict significance threshold yields too many long sequences and not enough short ones. The current program has some methods for suppressing the bogus long sequences, but does not find short sequences unless they compress very well.
Suppressing the incorrectly found long sequences can be done in several ways: by post-processing the output using various masking criteria [4], by using a better null model, or by insisting that the average savings per base is large (say at least 0.1 bits/base).
Since the standard deviation of the cost of a random sequence of
length n grows with ![]() , the threshold should probably have
the form
, the threshold should probably have
the form ![]() , rather than being a simple constant, but it
is unclear how to set a and b to get a given significance level
for a database.
, rather than being a simple constant, but it
is unclear how to set a and b to get a given significance level
for a database.
A significant sequence is present when the greatest savings seen is greater than T, but the scan is continued until either we reach the end of the cost map or the savings per position drops to less than half the savings per position at the point of greatest savings. The significant sequence runs from the start of the scan to the location of the greatest savings.
This method finds sequences that are significant, but the endpoints may not be optimally chosen. If we just picked the maximal segment (as in [6]), we would have extraneous characters on the ends of the segments. If the model for the interesting sequences provides estimates for uninteresting positions that are as good as the null models, then the savings for a junk character is zero or slightly positive (violating the requirements of a scoring system used with a maximal-segment method). In fact, early versions of the program reported the maximal segments and picked up long strings of junk.
One way to correct the problem would be to trim the significant sequences to maximize the savings per position. This approach, however, is too conservative, throwing away parts of the sequence that remain interesting.
Instead, the program tries to maximize the signal-to-noise ratio
(SNR).
The noise (the standard deviation of the savings for an uninteresting
sequence) should grow with ![]() for random sequences of length
n, so maximizing
for random sequences of length
n, so maximizing
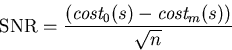
We can move the endpoints inward one position at a time, keeping track of
the position that gives the maximum SNR.
To make sure we do not lose any significant sequences, we stop the
trimming before the remaining savings would be less than ![]() .
.
After finding a sequence, we can restart the scan at the right
endpoint of the reported sequence.
Restarting here will scan some bases repeatedly, which may seem like a
violation of the assumption used for determining the significance
threshold, but the only way to find a sequence containing one of these
rescanned positions is for the position to be part of two sequences
that each saves at least the threshold.
This doubled savings is great enough that it would be significant even if all
![]() subsequences had been examined.
subsequences had been examined.
The strengths of a simple Markov model are that it allows very fast searching (the cost of each position can be computed in constant time), it has fairly small memory demands (4k+1 words for an alphabet of four letters), and the model can be built quickly from a fairly small seed.
The weaknesses are that the models are not human-readable and are not directly useful for aligning sequences. Furthermore, any extraneous junk that is in the set of seed sequences is included in the model--not just the interesting part of the seed. This can also be a strength, when the sequences are not well understood, since the neighboring bases may turn out not to be extraneous after all.
Some previous work with Markov models has concentrated on using them to predict the frequencies of short words [12,2,15]. These studies have generally found that fairly low-order models (k=2 to k=4) trained on the entire database work best for that application. In contrast, for searching we use fairly high-order Markov models (k=6 to k=10) and small seeds (as low as 30 characters). Even with a large seed of 12,000 characters (essentially all the REPs), the average value of a count for the 49 words of an order-8 model is only 0.046. With such small seeds and large models, almost all contexts have zero counts for all four characters, making the way zero counts are handled particularly important. For proteins, there are several regularizers for handling zero counts that work well [8], but for DNA we need to use a somewhat different approach.
High-order Markov models provide much more selectivity than low-order ones, but the small counts that result from training can make them too sensitive to noise. One way to reduce this problem is to add a small amount to the counts of the high-order model based on the probabilities assigned by a simpler model. The zero-offset technique is the simplest example of this, where the prior model just assigns identical probabilities to all possible letters. The program allows an arbitrary model m to be used, with weight wm.
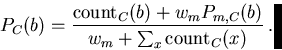
Using a Markov model with order about half the order of the final model produced noticeable improvements in the number of bits needed to encode the set of REPs. (The simpler model in turn used a flat prior.)
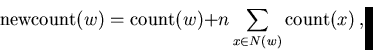
The program uses a slightly more sophisticated model, with three
blurring parameters n1, n2, and n3.
The n1 weight is used for words whose difference is
(A![]() G) or (C
G) or (C![]() T).
The n2 weight is used for words whose difference is
(A
T).
The n2 weight is used for words whose difference is
(A![]() C) or (G
C) or (G![]() T), and n3 is used for
(A
T), and n3 is used for
(A![]() T) or (G
T) or (G![]() C).
C).
The neighbor weights are best chosen by optimizing the adaptive compression of a related set of sequences, using a simple optimization technique, such as gradient descent, on the four parameters wm, n1, n2, and n3. The optimal value of wm is much smaller when neighbor blurring is used, since the neighbor blurring eliminates many of the zero counts, and provides a more accurate estimate of the probabilities than the simple prior model.
In many cases, we want to look for sequences on either strand of the DNA. We can achieve this by complement blurring of the word counts:
![]()
Complement blurring and neighborhood blurring can be combined:
The program actually uses three neighborhood blurring parameters (as described in Section 2), but applies the same blurring parameters in both the complemented and uncomplemented neighborhoods.
One other useful property of simple Markov models is that they can be used to do adaptive compression--that is, the model can be trained on the database up to, but not including, the base being scored. Initially, the model predicts uniform probabilities, and gradually adapts to the actual probabilities in the database. Adaptive compression gives us a useful way to determine how well a model generalizes, since it is being tested for many different training set sizes.
When choosing parameters for a model, we can optimize them for adaptive compression of a set of seed sequences. The minimization is currently done by using Newton's method to set the derivative of the encoding cost to zero, but almost any standard optimization technique should work. If the relationships between the seed sequences are similar to those of the sequences we will use the model to find, then this parameter setting works very well.
| order | n1 | n2 | n3 | c | wm | bits | bits/base |
| 0.0000 | 0.0000 | 0.0000 | 0.0000 | 396.656 | 24908.5 | 2.0012 | |
| 1 | 0.0000 | 0.0000 | 0.0097 | 1.8299 | 67.485 | 24489.6 | 1.9675 |
| 2 | 0.0069 | 0.0000 | 0.0000 | 0.9247 | 11.197 | 24108.0 | 1.9368 |
| 3 | 0.0156 | 0.0045 | 0.0000 | 0.7476 | 4.116 | 23376.1 | 1.8780 |
| 4 | 0.0147 | 0.0093 | 0.0040 | 0.7761 | 1.871 | 23105.5 | 1.8563 |
| 5 | 0.0395 | 0.0176 | 0.0069 | 0.6600 | 1.274 | 23012.6 | 1.8489 |
| 6 | 0.0589 | 0.0254 | 0.0178 | 0.5243 | 0.714 | 22653.2 | 1.8200 |
| 7 | 0.0781 | 0.0410 | 0.0277 | 0.4915 | 0.409 | 22006.6 | 1.7680 |
| 8 | 0.1005 | 0.0661 | 0.0557 | 0.4988 | 0.257 | 21429.6 | 1.7217 |
| 9 | 0.1465 | 0.0947 | 0.0705 | 0.5393 | 0.201 | 21041.8 | 1.6905 |
| 10 | 0.2055 | 0.1315 | 0.1249 | 0.5052 | 0.157 | 20885.1 | 1.6779 |
| 2 | 0.0045 | 0.0000 | 0.0000 | 0.9076 | 18.176 | ||
| 1 | 0.0000 | 0.0000 | 0.0097 | 1.8299 | 67.485 | 24116.2 | 1.9375 |
| 4 | 0.0175 | 0.0141 | 0.0073 | 0.7209 | 3.631 | ||
| 2 | 0.0069 | 0.0000 | 0.0000 | 0.9247 | 11.197 | 23310.9 | 1.8728 |
| 6 | 0.0548 | 0.0241 | 0.0182 | 0.5152 | 1.056 | ||
| 3 | 0.0156 | 0.0045 | 0.0000 | 0.7476 | 4.116 | 22635.5 | 1.8186 |
| 8 | 0.0919 | 0.0542 | 0.0465 | 0.4749 | 0.447 | ||
| 4 | 0.0147 | 0.0093 | 0.0040 | 0.7761 | 1.871 | 21365.7 | 1.7165 |
| 10 | 0.2088 | 0.1141 | 0.1124 | 0.4463 | 0.416 | ||
| 5 | 0.0395 | 0.0176 | 0.0069 | 0.6600 | 1.274 | 20415.6 | 1.6402 |
Table 1 gives a table of the parameter setting for the best adaptive compression of the 125 sequences (12477 bases) of high-m8-4-n0-i4-i1-e1, both for single-level models and for two-level models.
There is a difference between the true compression cost of a subsequence and the cost from the map for simple Markov models, but the only difference in cost is in the first k positions, since there is no compression for the first k positions of a sequence. If the cost map finds a low cost starting in position i, then we can compensate for the startup error by reporting the sequence starting at i-k. The program then recomputes the cost using the model for this lengthened subsequence, but this recomputation is probably not necessary, as it makes very little difference.
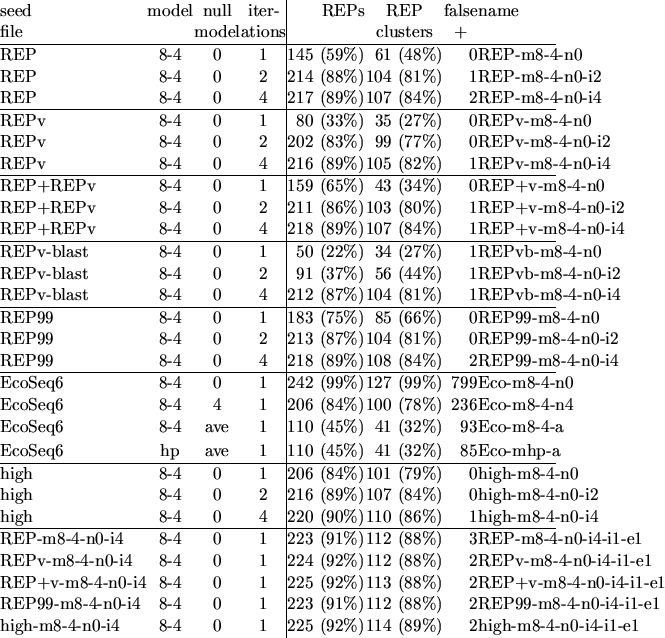
As an example of the techniques described in the preceding sections, the cost map method was used to find clusters of repetitive extragenic palindromic sequences (REPs) in the 1,875,932 bases of the EcoSeq6 database [13]. The sequences found were compared with two lists maintained by Ken Rudd [14]: one that contains 244 REPs (7474 bases) and one that groups them into 128 clusters (10501 bases). Each of these lists has some sequences not included in the other--sequences reported as false positives here occur on neither list.
The three search techniques used for building Rudd's list were described and referenced in Table I of [5]. The best of the techniques mentioned there (self-BLAST) found 106 of 112 REP clusters in EcoSeq5 or about 95%. However, many of the newer REPs in the EcoSeq6 list were provided by different sorts of searches, and it is not clear what percentage of the EcoSeq6 list would be found by self-BLAST. The self-BLAST method is quadratic in the size of the database, unlike this method and the one in [10], both of which are linear in the size of the database. Although individual BLAST searches with short query sequences are quite fast, the 448 searches needed for self-BLAST take a long time (about 100 times longer than an iteration of the search method presented in this paper, not counting the time needed for post-processing the voluminous self-BLAST output into a list of REPs).
Since there is, as yet, no biological test for REPs, it is difficult to decide how distant a match should still be considered part of the set. Rudd's list includes some that are quite far from the consensus sequence and excludes others that seem to be closer.
The current consensus sequence for a REP [5] is
A variant on the REPs, named REPv, has also been identified, and given
the following consensus sequence [14]:
It would be interesting to compare these searches with a standard search tool like BLAST [1], but BLAST can only search for matches to a single query sequence, and the REP consensus sequence has too many wildcard characters for it. A BLAST search was done with the REPv consensus sequence as a query, and the parameters set to allow very distant matches (W=8 and E=10.). This search found 68 sequences, some quite short. If E had been set to 0.01, as it is for the searches reported here, only 30 sequences would have been found.
There are many variables that can be adjusted in the Markov model searches: the seed chosen, the structure and parameters of the model, the null model, and the significance parameter E.
Various seeds were tried, including the REP consensus sequence, the REPv consensus sequence, both consensus sequences, the REP cluster REP99, and the 68 sequences found by BLAST from REPv. All worked reasonably well, though larger seeds generally found more sequences (see Table 2).
The two-level model in Figure 1 was used as the model--the parameters were chosen somewhat arbitrarily to provide fairly substantial blurring and to accept REPs on either strand of the DNA. The null model was a zero-order model trained on the entire database--the same null model used by most search programs. More sophisticated null models were tried, such as a 4th-order Markov model trained on the entire database, but these occasionally produced false positives from regions that were not well-modeled by the null model.
 |
We can increase the sensitivity of the test by using the results of the search as an expanded seed, retraining the model on this seed, and searching again (usually with the same null model). Since the initial model has somewhat arbitrary parameters, the program reoptimizes the model after each iteration, picking parameters that give the lowest cost for adaptive compression of the sequences found on that iteration. The main effect of this re-optimization is to reduce the blurring parameters substantially. Once we have trained on many representatives of the family we're looking for, there is less need to generalize the model by heavy blurring. The null model is not changed from one iteration to the next.
The basic technique for finding repeated patterns is to train a model on the entire database, then look for regions of the database that have particularly low encoding costs. To avoid the problem of reporting tandem repeats, the model is ``un-trained'' on each sequence of the database before searching that sequence. This technique increases the selectivity so that only subsequences similar to subsequences in other parts of the database are reported. Since training and un-training both take time linear in the length of the training sequences, we can still search the entire database in time proportional to the length of the database.
For these tests, the model was the two-level model in Figure 1. If a zero-order model is used for the null model, over 93% of the EcoSeq6 database is found--clearly this null model is too weak. Even if an order-4 null model (no blurring) is used, still over 55% of the database is found.
Another null model to use is a fake model that provides a constant ``probability'' for each character, based on the average compression obtained by the model on the whole database. For example, the model of Figure 1 trained on the whole EcoSeq6 database can encode it in 1.90917 bits/base. Using this average encoding cost as the null model, the program finds 135 sequences, totaling 139,751 bases, only about 7% of the database. Many of the sequences found are easily recognizable as known repeating units (REPs, IS5, IS2, IS30, ... ). Although only about 3% of the bases found are in REPs, 42 (31%) of the sequences are REPs. Some of the other repeats are much longer than the REPs, but much less frequent.
A model that gave a high weight to a flat prior should be more selective for high-frequency repeats, since low and moderate frequency repeats would not be distinguished as much from the background. Repeating the concentration of EcoSeq6 using a model with the same parameters as in Figure 1, except with the prior weights increased by a factor of 10, finds 126 sequences totaling 142,233 bases. This set contains essentially the same REPs as the first model, and so represents no greater concentration.
Some of the REPs compress much better than the other sequences, probably because there are more copies of the REPs in the database to train on. If the sequences are sorted by the cost in bits per base, the sixteen lowest are all REPs. Taking these sixteen as a seed for an iterated search provides very good results, with about 90% of the REPs found, and only one false positive.
| start | stop | name | found |
| 173611 | 173633 | mrcBecoM-8812 | 5 |
| 720470 | 720518 | fur-ecoM-852 | 1 |
| 1122371 | 1122404 | msyBecoM-72 | 2 |
| 1564562 | 1564608 | sfcAecoM-2672 | 5 |
| 3660926 | 3660963 | uspAeco-841 | 5 |
| 4084504 | 4084528 | glnAecoM-2729 | 1 |
The sequence in mrcBecoM is just before REP7 and has excellent alignments with parts of 12 other REP clusters. The sequence in fur-ecoM is palindromic (at least for 26/34 bases), but only weakly similar to a sequence in REP13 (17/18 bases). The sequence in msyBecoM is just before REPv116a and is weakly similar to a region immediately after REP4 and a region in REP44c. The sequence in sfcAecoM is similar to one at the beginning of REP16a- and one in REP101b-. The sequence in uspAeco is very similar to ones in at least nine REP clusters. The sequence in glnAecoM is weakly similar to a region at the end of REP2a.
| name | sequence |
| mrcBecoM-8812 | ctggacggataaagcgtttacgc |
| fur-ecoM-852 | attaatacggcgtagacatgagtctacgccgcatcacatcaggcattga |
| msyBecoM-72 | gtaaaccggatgcggcttcgcaataaaaacgtag |
| sfcAecoM-2664 | ttgaaatcgtaaagcattgccggatg |
| uspAeco-841 | ttgttacctgatcagcgtaaacaccttatctggcctac |
| glnAecoM-2729 | gagcgactcacctgctccggcctac |
Some of the known REPs did not come up in any of the searches, and others rarely appeared. Most of the missed sequences are fairly far from the consensus sequence, but three of the misses are rather surprising: REP39-, REP60, and REP83. Some other REPs that surprisingly few searches caught are REP23-, REP66-, REP56-, REP3, REP127, and REP128-.
I have shown how cost maps can be used effectively to search for interesting DNA sequences using simple Markov models.
The cost-map search method can also be used with more sophisticated models, such as hidden Markov models (HMMs). These models violate the assumptions of cost-map search a little more strongly than simple Markov models, but if some care is used in constructing the models, they can be used effectively. See [7] for some early experiments on using cost-map search with HMMs and automatic construction of HMMs from search results--the HMMs there do an even better job of characterizing REPs.
There are other interesting repeated sequences found by concentrating EcoSeq6, but which are not easily separated out. For example, only one of the sequences found by Kunisawa and Nakamura [9] is in Eco-m8-4-a, and it is buried in the middle of a much longer sequence. Using that sequence as a seed finds their sequences, but also finds 41 REPs. Using their set of five examples as a seed for an iterative search finds a total of ten examples (8 of which were found in the search using the single long sequence). It would be useful to develop better techniques for finding moderately repeated sequences like these.
The current program is written for experimentation with different models, rather than high efficiency, and thus is quite slow: 18,000 bases per second--comparable in speed to the program in [10], which is also roughly linearly proportional to the size of the database. A previous, less flexible program using Markov models and cost maps searched at a rate of 52,000 bases per second on a Sparcstation 10, and 80,000 bases/second should be fairly easy to achieve with modest recoding. While not as fast as a simple BLAST search, this would still be quite respectable.
